Introduction
Analyzing data from RNA-sequencing (RNAseq) experiments is a frequent workflow in modern genomics. To help document my methods and to provide an overview for others, this guide explains the basics of the technology and the specifics of analyzing its outputs computationally.
What is RNAseq?
RNAseq is the bulk sequencing of RNA: the intermediary between DNA and protein. Through RNAseq, we can get a sense of which genes are expressed based on how much RNA is transcribed from a gene. Common applications include comparing gene expression levels in the presence and absence of treatments and constructing large gene expression panels to use in association studies. Note that RNAseq is nearly identical to DNA sequencing: we normally perform the additional steps of selecting polyadenylated RNAs (in eukaryotes) as well as converting our RNA to coding DNA (cDNA) via reverse transcription before sequencing.
Before the adoption of RNAseq, the conventional way of high-throughput gene expression measurement was through microarrays, chips with sets of specific RNA sequences attached that bind to RNAs of interest. RNAseq offers several advantages over microarrays, such as in cost, throughput, and the ability to profile organisms that have a yet-unsequenced genome (in fact, transcriptomes, or reference indexes of expressed RNAs, can even be assembled de novo from RNAseq datasets using tools like Trinity). Moreover, RNAseq also allows us to detect secondary features such as mutations, splicing, and allele-specific expression all through a single method of collection.
Next-generation sequencing (NGS), the most widely used process for nucleotide sequencing, is perhaps one of the most sophisticated technologies of our time, combining multiple breakthroughs in biochemistry, imaging, and information technology. This video by Illumina highlights the many steps involved in the sequencing by synthesis technique, which along with pyrosequencing and sequencing by ligation make up three main methods often considered as NGS. In essence, a typical RNAseq experiment works by capturing the RNA within a sample, breaking such RNA into fragments, and sequencing a predetermined length of the fragments. In paired-end sequencing, we sequence both ends; in single-end sequencing, we sequence only a single one.
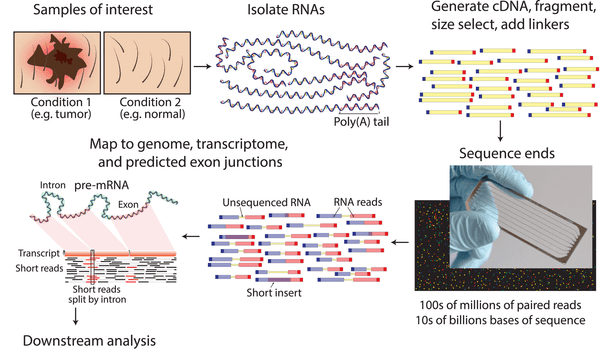
An overview of modern paired-end RNAseq. By Malachi Griffith, Jason R. Walker, Nicholas C. Spies, Benjamin J. Ainscough, Obi L. Griffith - PLoS Computational Biology, CC BY 2.5.
The computational problem
A typical RNAseq experiment will generate up to billions of sequenced reads distributed across the genome. This presents a challenging computational task: how can we map each of these reads to their correct locations in the genome in a way that is both timely and accurate? The human genome, for instance, is over three billion base pairs long. Furthermore, finding out where each read maps requires the consideration of imperfect matches caused by mutations and sequencing errors.
Besides these challenges of scale and fuzzy matching, we must keep in mind the fact that eukaryotic RNA is subject to splicing, through which parts are cut out by the cellular machinery as a mechanism of post-transcriptional regulation. When our reads originate from spliced RNA, they contain gaps when compared to the genome. Our algorithms should take these gaps into account and determine how best to match reads to the different transcripts that can originate from a single gene. Additionally, there is substantial variation in splicing junctions between samples, and in cancer we often discover novel junctions as well. Besides mapping our reads with known junctions in mind, our workflows should also have the capability to identify and incorporate new junctions from the samples at hand.
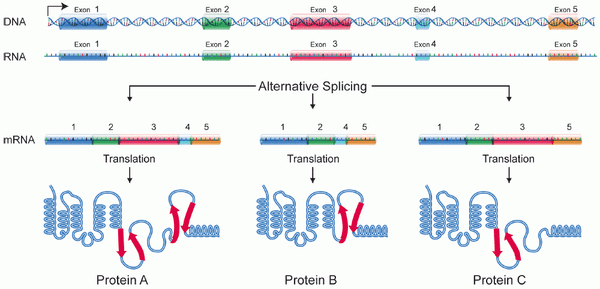
A schematic of RNA splicing. Through splicing, we see that the same DNA sequence may give rise to a variety of distinct RNA transcripts and thus different proteins. Source: National Human Genome Research Institute (NHGRI).
Tools of the trade
In this guide, I will walk through four tools: kallisto, which performs gene and transcript quantification; sleuth, which performs differential transcript analysis on the outputs of kallisto; STAR, which aligns reads to the genome as the first step in splicing estimation; and rMATS, which performs splicing quantification and differential analysis.
My choice of these three tools is not arbitrary. I selected them after examining and testing several alternatives because they satisfy several desired requirements:
-
Documentation and community: kallisto, sleuth, STAR, and rMATS are well-documented programs that have been cited hundreds of times – for instance STAR has been referenced over thirteen thousand times at the time of writing. Because of their wide usage, issues with these tools are likely to have solutions already posted on StackOverflow or Biostars, and online tutorials are similarly abundant.
-
Speed: Kallisto, STAR, and rMATS are extremely efficient programs. At the time of release, kallisto outperformed other programs by a factor of over 100, allowing jobs that once required a week's access to a dedicated computing cluster to be performed on a laptop in mere hours. Kallisto is efficient to the extent that the original paper found it to be I/O bound, or limited by the read/write speeds of the machine itself, in specific multicore settings.
STAR is also an extremely fast alignment algorithm. Provided you have enough RAM (about 48 GB minimum) at your disposal, it can finish most RNAseq runs in about an hour or so.
rMATS is also among the fastest splicing quantification programs. A typical splicing analysis workflow implementing STAR and rMATS can be completed in a few hours.
-
Load: Although this does not apply to STAR due to its high memory requirements, it does hold for kallisto and rMATS. As mentioned above, kallisto and rMATS can both process RNAseq experiments on a single laptop.
Kallisto
When kallisto was first published, the prevailing method for RNAseq quantification involved a combination of the TopHat2 (alignment) and Cufflinks (quantification) programs, both of which were relatively time-consuming and computationally intensive. Quantification required the expensive computation of read alignments to matching sequences in the genome or transcriptome, which was considered to be the rate-limiting step. Although improvements had been made using subsequence (or k-mer) matching with hash tables and streaming algorithms, such methods often come with tradeoffs in alignment accuracy.
Rather than align reads to the genome, the authors of kallisto introduced the concept of a pseudoalignment, a set of transcripts that can be matched to a read. By using a data structure called a de Bruijin graph (previously used in genome assembly) combined with k-mer hashing, kallisto delivered performance improvements of over 100x while simultaneously retaining accuracy and using substantially less memory. For a more thorough explanation, I recommend this blog post by Fong Chun Chan covering the concept of a pseudoalignment.
STAR
Unlike kallisto, which is considered to be "alignment-free," STAR does in fact align our reads to a reference genome. Compared to kallisto, STAR has a higher memory requirement and runtime, but the alignments it produces are required for several downstream applications such as quantifying RNA splicing, calling mutations, and identifying fusions. At the time of release, STAR was over 50x faster than competing aligners due to what the authors describe as a sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. This method translates to a two-step process:
-
Seed searching: First, STAR finds the longest sequence in the genome that perfectly matches some part of the read. Because of RNA splicing, discontinuous parts of the genome may map to the read. To account for this effect, we iteratively search for the unmapped regions in the read and match the next longest sequence until (nearly) the entire read is mapped. These discontiguous mapped regions are called the seeds, and they are formally referred to as maximal mappable prefixes.
Due to sequencing errors as well as mutations, we often cannot find perfect seed matches. When one part is unmapped, the previous seed is extended to include the mismatch. However, if the entire seed simply does not have a good match, the read is soft clipped and assumed to have a sequencing error.
-
Seed clustering and stitching: The separate seeds in a read are "stitched" to complete the original read. This merging is performed based on a predetermined scoring system for mismatches and gaps.
This bottom-up iterative process ultimately makes STAR much faster than its competitors, which instead attempt a top-down strategy of matching the entire read and then splitting. A more in-depth explanation and tutorial regarding STAR may be found in this lesson by the teaching team at the Harvard Chan Bioinformatics Core (HBC).
rMATS
rMATS takes the alignments from a program such as STAR and uses a statistical model to quantify the inclusion levels of exons as well as to compare these inclusion levels between sample groups. rMATS stands for replicate multivariate analysis of transcript splicing and was in fact based off an earlier version, MATS, that did not support experimental replicates (hence the lack of an r). The benefit of a statistical model capable of handling replicates is explained by the authors of the rMATS paper as follows:
Of note, pooling RNAs or merging RNA-Seq data from multiple replicates is not an effective approach to account for variability, and the result is particularly sensitive to outliers.
...
Moreover, certain commonly used experimental design or data analysis strategies in RNA-Seq studies of alternative splicing, such as pooling RNAs or merging RNA-Seq data from multiple replicates, are not recommended because they do not properly account for variability. Specifically, when the total sequencing count is fixed, the analysis of replicates always outperforms the analysis of a single RNA sample pooled from the individual replicates.
The specifics behind the hierarchical model used by rMATS are too detailed for this tutorial. If you are interested, they are explained in the original paper linked above.
Installation
Kallisto, STAR, and rMATS may each be installed by compilation of their source code, the instructions for which are detailed on their respective tutorials as linked.
Alternatively, kallisto and STAR are available as Anaconda packages, which may be easier to use in cluster settings. rMATS is also available as a Docker image, which is my personal preference because some of the dependencies (notably gfortran) may be tricky to install on macOS.
Expression analysis
The standard RNAseq expression workflow can be split into three steps: mapping, the assignment of reads to a reference genome/transcriptome; quantification, the estimation of the normalized read counts per gene and/or transcript, and differentiation, the comparison of quantifications between different experimental settings. Here, I will explain all three steps around a standard kallisto workflow.
Preparation
Before we start mapping any of our reads, we must first define an index that contains the transcripts that our reads should map to. The index is the stored representation of the de Bruijin graph that is key to kallisto's performance and accuracy. With kallisto, an index may be constructed using
kallisto index \
-i output_index.idx \
Homo_sapiens.GRCh37.75.cdna.all.faHere, we generate an output index called output_index.idx with the input sequences as Homo_sapiens.GRCh37.75.cdna.all.fa, or the version 75 release of the human transcriptome from ENSEMBL. This FASTA file may be obtained from here.
Note: the latest human genome build is now GRCh38, the latest transcriptome release of which is hosted here.
Quantification
Now that we have prepared our index, we can map and quantify our reads in a single command as follows:
kallisto quant -i output_index.idx \
--bias \
-b 100 \
-t 8 \
-o output_folder \
reads_end1.fastq reads_end2.fastqLet's walk through the arguments of this command.
- First, we use
--biasto tell the program to correct for biases in sequence content. This is caused by the fact that our ability to sequence RNA is confounded by factors such as gene length and various thermodynamic properties. - Second, we tell the program to generate 100 bootstrap samples with
-b 100. We need these for our differential analysis in the next section. If you do not want to do a differential analysis, this step can be skipped; in fact, it is often the most time-consuming step of the process. - Third, we specify the number of threads we want to use with
-t 8. Roughly speaking, this should be around the number of cores that your computer has (and double that if your processor supports hyper-threading). Using threads allows kallisto to perform pseudoalign our reads in parallel, leading to a large speed boost when possible. - Fourth, we use
-o output_folderto specify the folder for kallisto to save its outputs to. Note that we don't have to create the folder ourselves beforehand; kallisto will generate this directory itself. - Lastly, we pass in the name(s) of our sequencing read files with
reads_end1.fastq reads_end2.fastq, which should be in FASTQ format. In this case, our reads are in paired-end format, so we supply the files corresponding to each end. With single-end reads, we would only list a single file.
After completing, kallisto should produce the following outputs in output_folder:
abundance.h5: An HDF5 file containing bootstrapped transcript abundance estimates, required for subsequent differential analyses.abundance.tsv: A tab-delimited file of each indexed transcript and its estimated abundance in units of transcripts-per-million (TPM). See here for a thorough explanation of TPM and its alternatives by Lior Pachter, whose lab developed kallisto.run_info.json: A JSON file with run metadata such as the exact command used, start time, number aligned, etc. This file can be useful for diagnosing faulty runs or for documentation later on.
Differentiation
A major benefit of kallisto is the fact that the very same authors also developed a package, sleuth, for computing differential analysis on its outputs. Besides the benefits of vertical integration, sleuth is itself a good tool for differential analysis – at the time of publishing, it significantly outperformed existing methods in accuracy. Moreover, sleuth allows for complex combinations of conditions and covariates to be statistically accounted for, and it is also well-documented with a large user base.
To use Sleuth, we must first install its R package:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("pachterlab/sleuth")Once this step is completed, we can load it accordingly:
library(sleuth)Now, we feed sleuth information about our experiment setup as follows. With sleuth, the samples and conditions are stored in a table along with a column indicating the path to the kallisto output folder. Here, we will use the basic example of two conditions each with three replicates: a control group with triplicates control_1, control_2, and control_3; and a treatment group with triplicates treatment_1, treatment_2, and treatment_3.
Our table is therefore as follows:
| sample | condition | path |
|---|---|---|
| control_1 | control | control_1 |
| control_2 | control | control_2 |
| control_3 | control | control_3 |
| treatment_1 | treatment | treatment_1 |
| treatment_2 | treatment | treatment_2 |
| treatment_3 | treatment | treatment_3 |
Assuming we have specified this table in experiment_setup.txt, we can load as follows:
experiment_setup <- read.table(file.path("../experiment_setup.txt"),
header = TRUE,
stringsAsFactors = FALSE)
condition_name <- "treatment" # set the name of the condition to be compared (can also be "control")By default, sleuth computes differentially expressed transcripts rather than genes. If we want to aggregate these to get a differential expression analysis over genes, we can first use biomaRt to first fetch transcript-to-gene mappings:
library("biomaRt")
mart <- biomaRt::useMart(biomart = "ENSEMBL_MART_ENSEMBL",
host = "feb2014.archive.ensembl.org",
dataset = "hsapiens_gene_ensembl")
t2g <- biomaRt::getBM(attributes = c("ensembl_transcript_id",
"ensembl_gene_id",
"ensembl_peptide_id",
"hgnc_symbol",
"entrezgene",
"transcript_biotype"),
mart = mart)
t2g <- dplyr::rename(t2g,
target_id = ensembl_transcript_id,
ens_gene = ensembl_gene_id,
hgnc_gene = hgnc_symbol,
entrez_gene = entrezgene)
t2g <- dplyr::select(t2g,
c('target_id',
'ens_gene',
'hgnc_gene',
'entrez_gene',
'transcript_biotype'
))
t2g$duplicate_transcript <- duplicated(t2g$target_id)
t2g <- t2g[which(!t2g$duplicate_transcript),]
write.table(t2g,
file = "../sleuth_diff/ensembl_t2g.csv",
sep = ",",
col.names = TRUE,
row.names = TRUE)Now, we prepare our sleuth object by specifying the transcript-to-gene mapping through the target_mapping attribute:
so <- sleuth_prep(setup,
extra_bootstrap_summary = TRUE,
read_bootstrap_tpm = TRUE,
target_mapping = t2g,
aggregation_column = 'entrez_gene')Next, we fit the models, after which we run likelihood ratio tests to calculate significance:
so <- sleuth_fit(so, ~condition, 'full')
so <- sleuth_fit(so, ~1, 'reduced')
so <- sleuth_wt(so, condition_name)To extract our gene-level results, we specify pval_aggregate=TRUE in our call to sleuth_results to combine transcript-level significance estimates into gene-level ones:
table <- sleuth_results(so,
condition_name,
'wt',
show_all = TRUE,
pval_aggregate = TRUE)
write.table(table,
file = "sleuth_gene_results.csv",
sep = ",",
col.names = TRUE,
row.names = TRUE)To extract our transcript-level results, we specify pval_aggregate=FALSE in the same call:
table <- sleuth_results(so,
condition_name,
'wt',
show_all = TRUE,
pval_aggregate = FALSE)
write.table(table,
file = "sleuth_transcript_results.csv",
sep = ",",
col.names = TRUE,
row.names = TRUE)Note that sleuth is capable of powerful comparison functionality beyond the simple example presented here. For instance, covariates and batch effects are also able to be incorporated into the model. A more detailed tutorial on Sleuth by the developers can also be found here in addition to a list of official walkthroughs. For those interested in the mechanics of the statistical model, I recommend checking out this post by Lior Pachter.
Splicing analysis
Beyond bulk gene/transcript expression, a common mode of analysis is to also look at the levels of spliced sequences in expressed RNAs. To look at RNA splicing, we must first compute alignments to the genome. For this step, we will use STAR, after which we will use rMATS to perform quantification and differential analysis.
The workflow for STAR is pretty similar to that of kallisto: we first assemble an alignment index based on our genome and/or transcripts, and we subsequently use this index to match reads to genomic regions. Once STAR is completed, the outputs can immediately be fed into rMATS, which computes splicing measurements and performs differential analyses.
Preparation
Index generation with STAR is handled by a single command:
STAR --runMode genomeGenerate \
--runThreadN 8 \
--genomeDir STAR_index \
--genomeFastaFiles Homo_sapiens.GRCh37.75.dna_sm.primary_assembly.fa \
--sjdbGTFfile Homo_sapiens.GRCh37.75.gtf \
--sjdbOverhang 100Let's walk through the arguments of this command:
- First, we use
--runMode genomeGenerateto specify that we are generating an index (differentrunModearguments handle alignment and other operations provided by STAR). - Second, we use
--runThreadN 8to specify that the program will use eight computing threads. - Third, we use
--genomeDir STAR_indexto tell the program to output all of the index files toSTAR_index. - Fourth, we use
--genomeFastaFiles Homo_sapiens.GRCh37.75.dna_sm.primary_assembly.fato point the indexer to the reference FASTA file, which in this example is the human genome. Specifically, our FASTA file contains the 75th ENSEMBL release of human genome version GRCh37, which may be found here. - Fifth, we use
--sjdbGTFfile Homo_sapiens.GRCh37.75.gtfto specify the splice junction annotation file that we will use, which should be in GTF format. In this case, we use the corresponding GRCh37.75 GTF file matching our earlier--genomeFastaFilesargument. This GTF file is hosted on ENSEMBL here. - Lastly, we use
--sjdbOverhang 100to define the splice junction overhang. This value ideally should be set to the read length minus one, but according to the manual, a value of 100 (the default) should be just as good as the ideal.
Note that STAR index construction, like STAR alignment, is a very memory-intensive step. Indexing the human genome requires about 48 GB of RAM, so this step should be done on a computing cluster unless you have a powerful enough workstation.
Alignment
Once we have our STAR index, we can perform alignment in a single command:
STAR --runMode alignReads \
--genomeDir STAR_index \
--readFilesIn reads_end1.fastq reads_end2.fastq \
--outFileNamePrefix output_dir \
--outSAMtype BAM SortedByCoordinate \
--twopassMode Basic \
--twopass1readsN 1000000000000 \
--sjdbOverhang 100 \
--runThreadN 8The arguments of this command are as follows:
- First, we use
--runMode alignReadsto specify the STAR submodule we want: in this case, we want to align reads. - Second, we use
--genomeDir STAR_indexto point our run to the index that we generated previously. - Third, we use
--readFilesIn reads_end1.fastq reads_end2.fastqto specify the paired-end FASTQ files that we want to align (as with kallisto, we would only specify one FASTQ file for single-end reads). - Fourth, we use
--outFileNamePrefix output_dirto specify our desired output directory. This will contain our alignment files as well as several progress reports produced while the program runs. - Fifth, we use
--outSAMtype BAM SortedByCoordinateto specify the type of output alignment file that we want. In this case, we want a BAM, or Binary Alignment Map, which is a compressed representation of our aligned reads (the uncompressed version is called SAM, or Sequence Alignment Map). The last part tells STAR to sort our alignment by the genomic coordinate, which is often required by downstream methods such as rMATS. Note that the alignment file may also be manipulated afterward using the samtools package. - Sixth, we specify
--twopassMode Basicto tell STAR to actually align our reads twice. The reason for this optional second pass is that standard single-pass STAR can often fail to account for splice junctions that weren't provided in our original index, which makes it suboptimal for purposes such as estimating splicing and/or mutations. This can be a particular drawback in cancer samples, which typically contain many uncataloged mutations and splice sites. Here, we detect and collect novel splice junctions in the first step and reindex before realigning in the second. - Seventh, we use
--twopass1readsN 1000000000000to tell the program to pass all reads through the first alignment step. This parameter is intended to limit how many reads we first align for purposes of speed, but in this case, we set it to an absurdly high number to align them all. - Eighth, we use
--sjdbOverhang 100with the same purpose as explained in indexing. Note that the value provided here must match that of the index; otherwise an error will be raised. - Lastly, we specify with
--runThreadN 8to specify the number of computing threads we want to use.
After finishing (and while running), STAR should produce the following outputs in output_dir:
Log.out,Log.progress.out,Log.final.out: Log files for quality assurance.SJ.out.tab: A list of detected splice junctions and read counts. Useful for custom splicing analysis, which we ended up using in the CCLE2 study.Aligned.sortedByCoord.out.bam: The sorted-by-coordinate BAM alignment file. Based on your arguments to--outSAMtype, the exact name of this file may differ – for instance, here we have abamfile that has been sorted by coordinate, hence thesortedByCoordinfix._STARpass1/: A temporary directory for the first-pass alignment that retains the splice junctions and logs for debugging._STARtmp/: A temporary directory used by STAR for BAM sorting and other alignment file postprocessing steps._STARgenome/: A directory containing the detected splice junctions used for two-pass alignment.
rMATS
Having computed alignments with STAR, we are now ready to quantify splicing events with rMATS. In this example, I will use the Docker implementation of rmats:turbo01, further documentation for which is available here.
Before running rMATS, we need to prepare two files detailing the samples we want to compare (if you are not performing a differential analysis, you can determine these arbitrarily). These are comma-delimited files, so for instance one containing three STAR outputs might look like this:
STAR_output_1/Aligned.sortedByCoord.out.bam.BAM,STAR_output_2/Aligned.sortedByCoord.out.bam.BAM,STAR_output_3/Aligned.sortedByCoord.out.bam.BAMNote the fact that the commas are not followed by spaces.
Quantifying and performing differential analyses with rMATS is now accomplished in a single command:
docker run -v DATA_PATH:/data rmats:turbo01 \
--b1 control_samples.txt \
--b2 treatment_samples.txt \
--gtf Homo_sapiens.GRCh37.75.gtf \
--od rmats_output_dir \
-t paired \
--nthread 8 \
--tstat 8Let's dissect this command:
- First, we mount the directory containing all our files to our Docker runtime with
docker run -v DATA_PATH:/data rmats:turbo01. In this case, our directory isDATA_PATH. All other files referenced in this command will need to be underDATA_PATHand referenced as ifDATA_PATHis the root directory. - Second, we use
--b1 control_samples.txtto specify the file listing the first group of samples, which in this case is our controls. - Third, we use
--b1 treatment_samples.txtto specify the file listing the second group of samples, which in this case is our treatments. - Fourth, we use
--gtf Homo_sapiens.GRCh37.75.gtfto specify a GTF file containing our transcripts, which specify our splice junctions. - Fifth, we use
--od rmats_output_dirto define our desired output directory, or where rMATS will write all of our results to. - Sixth, we use
-t pairedto notify rMATS that we are working with paired-end reads. - Seventh, we use
--nthread 8to specify that we want to use eight threads. - Lastly, we use
--tstat 8to specify that we also want to use eight threads in the statistical model.
After running, rMATS will produce a bunch of files in rmats_output_dir. rMATS defines five categories of splicing events: A3SS (alternative 3' splice site), A5SS (alternative 5' splice site), SE (skipped exon), IR (intron retention), and MXE (mutually exclusive exons).
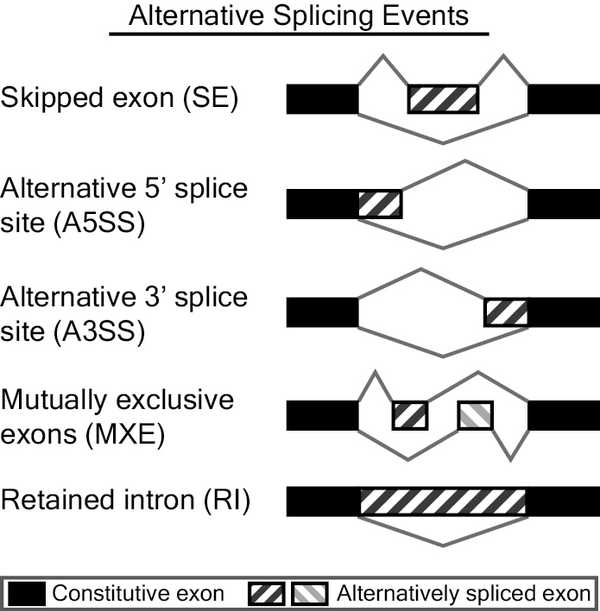
Schematic of the splicing categories used by rMATS. Source: rMATS website.
For each splice type, rMATS then produces the following:
<SPLICE_TYPE>.MATS.JC.txt: Splicing quantification and comparisons using the junction reads that span the splice junctions.<SPLICE_TYPE>.MATS.JCEC.txt: Splicing quantification and comparisons using both junction reads and reads covering the targeted exon. JC and JCEC are almost always consistent, although one might prefer JCEC for having more reads.fromGTF.<SPLICE_TYPE>.txt: Splicing events from the GTF annotation file as well as novel detected events.fromGTF.novelEvents.<SPLICE_TYPE>.txt: Only novel detected events.JC.raw.input.<SPLICE_TYPE>.txt: Inclusion and exclusion counts using only junction-spanning reads for each splicing event.JCEC.raw.input.<SPLICE_TYPE>.txt: Inclusion and exclusion counts using junction-spanning and target exon-containing reads for each splicing event.
This concludes our splicing quantification and analysis workflow. The outputs from rMATS, which are tab-delimited, can now be loaded and visualized using a program/framework of your choice – for instance, Python, R, or even Excel. For more details on installation and testing, see the rMATS user guide.
Additional tips
Where can I find RNAseq data?
Studies frequently reanalyze existing RNAseq runs from previous experiments, often to validate new hypotheses as well as to apply or benchmark new methods. Several governments and nonprofits maintain a variety of open-access repositories with data from RNAseq and beyond:
- The Sequence Read Archive (SRA) is provided by the National Center for Biotechnology Information (NCBI) and is probably the most comprehensive sequencing repository out there. I recommend using tools such as Aspera and SRA explorer to download files, as the default tools provided by NCBI are often slow.
- The European Nucleotide Archive (ENA) is organized by the European Bioinformatics Institute and contains many other projects not in SRA, although there is often overlap between the two. ENA files can also be downloaded using Aspera and SRA explorer.
- Large projects such as the Institute for Systems Biology Cancer Genomics Cloud (ISB-CGC) provide sequencing files for various cancer genomics projects such as the CCLE, TCGA, and others on Google Cloud Platform (GCP). Note that approval is required for working with raw sequencing files from projects such as CPTAC, TARGET, and TCGA due to patient confidentiality concerns.
- The Broad Institute hosts several genomics datasets on terra.bio including raw RNAseq runs as well as processed expression tables and other annotations. Terra can also be used to run workflows on GCP, either custom or selected from their library, provided you are can cover the costs. Note that raw read files for datasets are often under restricted access, as with the ISB-CGC.
What else can I do with RNAseq data?
RNAseq provides additional insights in addition to gene expression and splicing. Possible extensions may include:
- Mutations: As a copy of our DNA, mRNA also contains the same mutations and thus can be used for mutation calling. For instance, one study by the Genotype-Tissue Expression (GTEx) project developed and applied an RNAseq mutation pipeline across thousands of normal human tissue samples, finding that even our normal tissues are often rich in mutations that we typically associate with tumors.
- Allele-specific expression: Genes are often not expressed from both parental alleles, for instance in the phenomenon of genomic imprinting. By combining RNAseq mutation calling with DNA-sequencing, we can look at "anchor alleles" to find which genes are being expressed from a single one. This method has been used to look at X-chromosome inactivation across the GTEx dataset as well as to increase the power of genome-wide association studies.
- RNA editing: In certain scenarios, our mRNA undergoes adenosine-to-inosine (A-to-I) editing by ADAR (adenosine deaminase acting on RNA) enzymes, which can also be detected via RNAseq. The GTEx project has also released a paper in which they quantify and correlate this editing across their samples.
What other sequencing technologies are in use?
Next-generation sequencing is by no means the only technology available for DNA/RNA sequencing. In addition to the three mentioned in the introduction, there are a few others of particular note:
- Nanopore sequencing, which detects changes in electrical current as a DNA molecule passes through a protein pore, is capable of sequencing extremely long reads at low cost, but often with lower quality.
- Sanger sequencing, also called chain termination, was the method originally used to sequence the first human genome nearly two decades ago. Despite high costs compared to modern high-throughput sequencing, it is still in use today as a gold standard because of its high accuracy, and it is most often employed to confirm features detected by more modern techniques.
- Single-molecule real-time (SMRT) sequencing detects the activity of a single DNA polymerase enzyme extending the molecule of interest. Normally, this is done in massively parallel fashion, producing many reads at low accuracy that can be merged to form a stable consensus.
- Single-cell sequencing uses molecular barcoding techniques to allow us to discern individual cells in our samples. One particularly prominent application is single-cell RNAseq (scRNAseq), which kallisto is also capable of handling.
The Wikipedia page on DNA sequencing also provides more in-depth explanations as well as several other technologies.